
Présentation aux professionnels de santé
Avertissement : les informations suivantes sont destinées aux professionnels de la santé. Elles contiennent des termes techniques et médicaux spécifiques.
Définitions
Les porphyries sont un groupe de troubles de la voie de biosynthèse de l’hème présentant des symptômes neuroviscéraux, des lésions cutanées ou les deux. Toutes les porphyries proviennent d’un déficit partiel d’une des enzymes de la biosynthèse de l’hème (figure 1) et, excepté pour la porphyrie cutanée tardive, sont pour la plupart des maladies héréditaires monogéniques de transmission autosomique dominante. Le diagnostic clinico-biologique des porphyries nécessite l’identification des profils uniques de la surproduction des précurseurs de l’hème résultant de chaque déficit enzymatique (voir diagnostic).
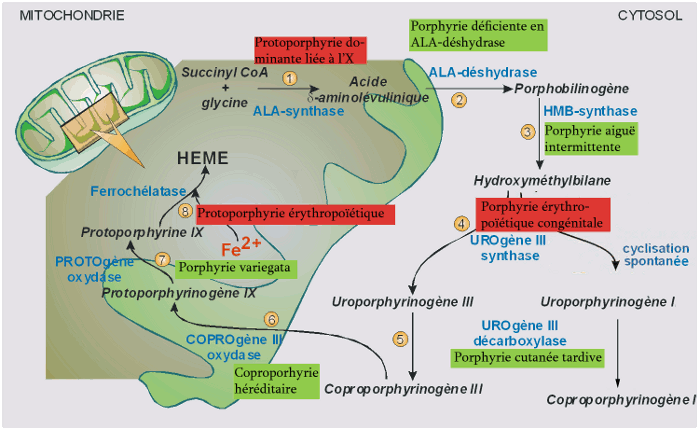
Figure 1 : biosynthèse de l’hème et porphyries associées.
| porphyries hépatiques | porphyries érythropoïétiques |
|---|
Toutes les porphyries autosomiques dominantes montrent une pénétrance clinique incomplète. La majorité (90% parmi les familles suivant les dernières estimations) de ceux ayant hérité d’une porphyrie reste asymptomatique durant toute leur vie mais on ne peut prédire quels individus entrent dans cette catégorie. Les termes latent ou présymptomatiques sont utilisés pour décrire les individus, quelque soit leur âge, n’ayant jamais eu de symptômes de porphyrie bien que présentant la mutation familiale. Le terme rémission est utilisé pour décrire les individus chez qui les symptômes de la porphyrie ont été résolus.
Porphyries aigües
- Porphyrie aigüe intermittente
- Porphyrie variegata
- Coproporphyrie héréditaire
- Porphyrie déficiente en δ aminolevulinate déshydrase
Les porphyries hépatiques aigües: la Porphyrie aigüe Intermittente (PAI), la Porphyie Variegata (PV) et la Coproporphyrie Héréditaire(CH)) sont caractérisées par des crises neuroviscérales aigües épisodiques qui peuvent exceptionnellement être fatales. Les crises aigües sont très rares avant la puberté, elles débutent habituellement entre 15 et 35 ans, principalement chez la femme. La majorité des patients ont une ou quelques crises, suivies par une rémission complète mais des crises récurrentes, qui peuvent parfois être liées au cycle menstruel chez la femme, apparaissent dans moins de 10% des cas. Les crises sont souvent déclenchées par les médicaments, l’alcool, les facteurs endocrinens, les infections ou les restrictions caloriques (jeûne).
La PAI est la plus fréquente des porphyries aigües. Sa prévalence est estimée à 1/10 000 en Europe(*).Les crises neuroviscérales sont les principales manifestations cliniques, les lésions cutanées n’apparaissant jamais. Elle est due au déficit partiel en activité de la PBGD responsable d’une accumulation de précurseurs.
Environ 60% des patients atteints de PV présentent seulement des lésions cutanées, 20% des crises neuroviscérales sans signe cutané et 20% les deux simultanément. La PV a une prévalence d’environ un tiers par rapport à la PAI dans la majorité des pays européens.
La CH est la moins commune des porphyries aigües. La majorité des patients présentent des crises aigües accompagnées dans 30% des cas de lésions cutanées. Les lésions cutanées seules sont rares dans la CH.
Cette PHA, ou porphyrie de Doss, est rarissime. Seulement sept patients ont été diagnostiqués en Europe depuis sa description en 1979. Tous ont développé des crises aigües neuroviscérales, parfois dès l’enfance, avec/ou des neuropathies périphériques.
Porphyries non aigües
- Porphyrie cutanée tardive
- Porphyrie érythropoïétique congénitale
- Protoporphyrie érythropoïétique
- Protoporphyrie érythropoïétique dominante liée à l’X
La PCT est une porphyrie cutanée (peau fragile, bulleuse, hypertrichose, pigmentation) qui peut être acquise (type I, 80% des cas) ou héritée (type II, 20% des cas) de façon autosomique avec une faible pénétrance. La majorité des patients ont des dommages sous-jacents des cellules hépatiques avec une surcharge en fer, très fortement associée avec l’alcool, l’hépatite C et les mutations du gène de l’hémochromatose (HFE).
La PEC est une affection autosomique récessive rare. Plus de 200 cas ont été diagnostiqués en Europe (*). La majorité des patients ont les symptômes peu après la naissance et ont une sévère photosensibilité, une érythrodontie et une anémie hémolytique. La greffe de moëlle osseuse est le seul traitement efficace.
La PPE est une maladie autosomique dominante avec une faible pénétrance. Sa prévalence est estimée à 9/10 000 en Europe(*). Chez la plupart des patients, l’expression clinique nécessite une co-transmission d’un allèle faible présent dans environ 10% de la population. C’est la seule porphyrie présentant une photosensibilité aigüe sans fragilité cutanée ni bulles, commençant dès la petite enfance. Les complications hépatiques, dûes à l’accumulation de protoporphyrines dans les hépatocytes, se produisent dans moins de 2% des cas.
La PPEDLX est une maladie dominante liée à l’X. Les signes cliniques sont exactement les mêmes que ceux observés avec la PPE.
Autres porphyries
Des variants rares des porphyries autosomiques dominantes surviennent dans lesquels des mutations dans les gènes du HMBS (PAI homozygotes), du CPO (harderoporphyrie et CH homzygote), de l’UROD (porphyrie hépatoérythropoïétique) ou de la PPOX (porphyrie variegata homozygote) ont été identifiés dans les deux allèles. Ils entraînent des symptômes dès la petite enfance et la majorité est plus sévère que chez leur équivalent hétérozygote.
(*) : Les cahiers de l’Orphanet – Prévalence des maladies rares : données bibliographiques – Novembre 2009 n°1